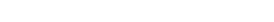İÇİNDEKİLER
- Önsöz
- Giriş
- Biyoteknolojik İlaçlar ve Üretim Süreçleri
- Biyobenzer İlaçlar ve Karşılaştırılabilirlik
- Biyobenzer Ürünlerde Ekstrapolasyon
- Biyobenzer İlaçlarda Değiştirilebilirlik
- Biyoüstünler
- Referans Ürünle Yeterli Karşılaştırma Çalışması Olmayan Biyolojik Ürünler
- Biyolojik ve Biyobenzer İlaçlarda Farmakovijilans, İzlenebilirlik ve Risk Yönetimi
- Yazarlar


BİYOLOJİK VE BİYOBENZER İLAÇLARDA FARMAKOVİJİLANS, İZLENEBİLİRLİK VE RİSK YÖNETİMİ
Farmakovijilans
Farmakovijilans “günlük klinik uygulamada ilaçların güvenliliği ile ilgili klinik verilerin toplanması, ilaç uygulamasında karşılaşılan sorunların takibi, sorumlu nedenlerin saptanması, tanınması, araştırılması, kaydedilmesi, duyurulması ve gerekli önlemlerin alınması” şeklinde tanımlanabilir.
Son yıllarda ilaç kullanımında yaşanan artış, ilaçlara bağlı sorunların daha fazla tartışılmasını beraberinde getirmiştir. Pazara yeni ilaçlar katılmakta, mevcut ilaçlarla ilgili deneyim genişlemekte, ilaçların etkililiği ve güvenliliği başta olmak üzere ilaçlar hakkındaki bilgiler ve görüşler sürekli değişmektedir.
Birçok ilacın bilinen yan etkilerine yenileri eklenmektedir. Saptanan yeni veriler ile ilaçlar için yeni endikasyonlar veya yeni kullanım yolları da ortaya çıkmaktadır.
Farmakovijilans açısından ilaçların güvenlilik profilleri büyük önem taşımaktadır. İlaçların güvenlilik profilleri ruhsatlandırma aşamasında detaylı olarak belirtilmelidir.
Ruhsatlandırma aşamasından önce yapılan ilaç geliştirme araştırmaları, ilaç güvenliliği konusunda oldukça yararlı bilgilerin ortaya çıkmasını sağlamasına karşın, bu konudaki endişelerin tam olarak giderilmesinde yetersiz kalmaktadır. Bunun nedenleri arasında ilaç güvenliliğiyle ilgili klinik öncesi çalışmalardaki hayvan deneylerinin sonuçlarının insandaki durumu her zaman yansıtmaması, klinik ilaç araştırmalarında ise kısıtlı sayıda, çoğunlukla seçilmiş hasta gruplarının çalışmalara dahil edilmesi, bu araştırmaların sınırlı zaman dilimlerinde yapılıyor olması, popülasyonun yaş, cinsiyet ve etnik köken yönünden sınırlandırılmış olması, komorbiditenin ve birlikte kullanılan ilaçların kısıtlanmış olması, ilaca maruz kalınan sürenin ve takip süresinin nispeten kısa olmasına bağlı sorunlar yaşanıyor olması sayılabilir (1).
Ruhsatlandırma sırasında neleri bilmiyoruz?
Günlük yaşamda ilacın etkililiği (effectiveness) ile ilgili olarak, uyunç, rezistans ve çalışılmayan popülasyonlar nedeniyle bilgi eksikleri bulunmaktadır.
İlacın tam güvenlilik profili için aşağıdaki tüm bilgilere ihtiyaç vardır:
- Nadir görülen advers reaksiyonlar
- Gecikmiş advers reaksiyonlar
- Kronik maruziyetten kaynaklanan advers reaksiyonlar
- Gebelikte ilaç kullanımı sonucu görülen advers reaksiyonlar
- İlaç etkileşimlerinden kaynaklanan advers reaksiyonlar
- Çalışılmamış popülasyonlarda görülen advers reaksiyonlar (çocuklar, yaşlılar, komorbiditeler)
- Suistimalden ve ilaç hatalarından kaynaklanan advers reaksiyonlar
- Endikasyon dışı kullanım sonucu meydana gelen advers reaksiyonlar
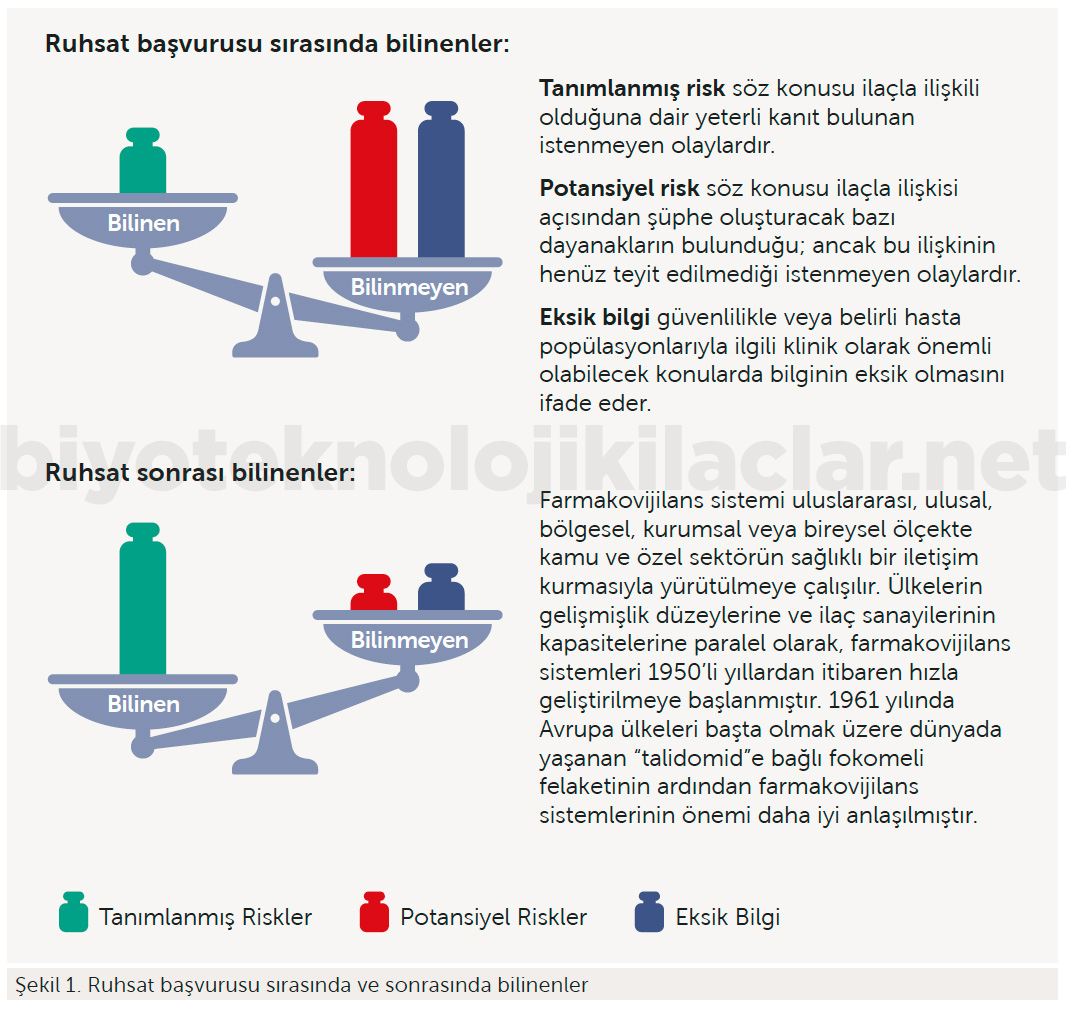
Pazarlama öncesi çalışmalardaki bu tür kısıtlılıklar, advers etkiler / olaylar bakımından ilaçların pazarlama sonrası izleminin önemini daha da artırmaktadır. İlaçların güvenliliğini pazarlama sonrasında izlemek, değerlendirmek, gerekli önlemleri almak ya da aldırmak, ulusal ve uluslararası yeterli iletişimi sağlamak için ulusal farmakovijilans sistemlerinin kurulmasına gereksinim duyulmuştur.
Farmakovijilans sistemlerinin kurulmasında ve bu alanda farmakoepidemiyolojik çalışmalar yapma konusunda, gelişmekte olan ülkeler bu sürecin gerisindedir. İlk bakışta, gelişmiş ülkelerin farmakovijilans verileri, farmakovijilans sistemine sahip olmayan bu ülkelerde de uyarlama yoluyla kullanılabilir gibi görülse de ilaç kullanımında kültürel ve genetik faktörler başta olmak üzere birçok yerel faktörün rolü, bu tutumun yanlışlığını ortaya koymaktadır. Örneğin ilaç kullanım sürecinde hasta uyuncu, hastaların ilaçlarının etkileri / yan etkilerini algılama biçimi, ilaca bağlı advers etki / olay geliştiğinde ne yapılacağı, polifarmasi alışkanlığı, ilaç-ilaç etkileşimi, ilaç-besin etkileşimi oranları ve çeşitliliği, hastaların, hastalıklarının hangi evresinde hekime gitme alışkanlıkları, ilaçlarla ilgili sorunların çözümünde bireylere bakış (yaş, cinsiyet, sosyal statü vb. faktörler) farklılıkları, ilaçlarla ilgili haber / yorum / reklamları dikkate alma alışkanlığı, değişik bölge ve kültürlerde kişilerin yaşam biçimleri, sigara, alkol, vb. kullanımı alışkanlıkları ve bunların ilaçlarla olan etkileşimleri, değişik bölgelerde bazı hastalıkların görülme sıklığının farklı olması, eczacı / ilaç dağıtıcılarının nakil, saklama kuralları ve ilaç son kullanım tarihlerine dikkat etme alışkanlıkları, ilaç alım gücü yönünden, hastaların ekonomik düzeyi veya geri ödeme kurumlarının tedavi giderlerini ödeme oranları, ilaçlara bağlı sorunların sağlık kuruluşlarına bildiriminde yaşanan sorunlar farmakovijilansı etkilemektedir. Hastalardan kaynaklanan farklılıkların dışında, düzenleyici otoritelerin alt yapı ve işleyiş farklılıkları, hekimlerin ve diğer sağlık personelinin advers etki / olay bildirimindeki bilgi ve tutumları, ilaç endüstrisinin periyodik yarar risk değerlendirme raporlarının (PYRDR) bildirimleri başta olmak üzere iyi farmakovijilans uygulamaları konusundaki farklılıkları ve yukarıda sayılan tüm faktörler, ulusal farmakovijilans sisteminin gerekliliğini açık bir şekilde ortaya koymaktadır.
Ulusal Farmakovijilans Sistemi
Ülkemizde, Beşeri İlaçların Güvenliğinin İzlenmesi ve Değerlendirilmesi Hakkında Yönetmelik” 22 Mart 2005 tarih ve 25763 sayılı Resmi Gazete’de yayımlanarak “Beşeri İlaç Ruhsatı Sahipleri için Farmakovijilans Kılavuzu” ile birlikte 30 Haziran 2005 tarihinde yürürlüğe girmiştir. Farmakovijilans Yönetmeliği 15 Nisan 2014 tarihinde yenilenmiştir.
Ulusal farmakovijilans sisteminin hedefi; pazarlama sonrası, klinik kullanımda olan ilaçların risk / yarar oranlarının bireysel ve toplumsal düzeyde mümkün olabilecek en iyi duruma getirilmesini sağlamaktır.
Ulusal farmakovijilans sistemi, yeni ruhsatlandırılmış ya da ciddi advers etkileri güncel tartışma konusu olan ilaçlar başta olmak üzere, tüm ilaçların advers etkilerini tespit etmek için kanallarını açık tutmak zorundadır. Veri toplamak için gerekirse bu konuda özgün bilimsel araştırmalar yapılmasını talep edebilir, yeni stratejiler belirleyebilir. Özellikle immünojenisite sorunu tartışılan ilaçlarla ilgili pazarlama sonrası gözlemsel çalışmaların yapılmasını teşvik ederek referans veya biyobenzer ürünle ilgili sorunların fark edilebilirliğini ve saptanmasını kolaylaştırabilir, ulusal ölçekli veri tabanı oluşturabilir. Zira advers etkilere bağlı ortaya çıkan sorunların bildirimlerindeki yetersizlik tüm dünyanın sorunu olduğu gibi ülkemiz için de yeterli düzeyde değildir.
Spontan advers reaksiyon bildirimleri farmakovijilans sisteminin temel unsurudur. Advers reaksiyon bildirimleriyle aktarılan veriler sinyal tespit faaliyetleri için önemli bir kaynak niteliği taşımaktadır. Sağlık Bakanlığının Stratejik plan doğrultusunda hazırlanan 2019 yılı Performans Programı hedefleri çerçevesinde yürütülecek faaliyetlerden biri de İlaçların güvenli kullanımına katkı sağlamak amacıyla farmakovijilans ile ilgili farkındalığın ve buna bağlı olarak advers reaksiyon bildirimlerinin artırılmasını amaçlamıştır.
Ülkemiz nüfusuna göre yılda milyon kişi başına düşen bildirim sayısı, Farmakovijilans sisteminin kurulduğu 2005 yılından bu yana anlamlı bir ivmeyle artmışsa da nüfusumuz göz önüne alındığında oldukça düşük olduğu Şekil 3’teki gibi sunulmuştur (2). Zira, Dünya Sağlık Örgütü yılda milyon kişi başına düşen bildirim sayısının en az 200 olmasını önermektedir. Nitekim, gelişmiş ülkelerde advers reaksiyon bildirim sayıları bir milyon nüfus başına yılda en az 250 bildirim şeklindedir. Dünya Sağlık Örgütü Uluslararası İlaç İzleme İş birliği Merkezine Üye ülkeler arasında karşılaştırma yapılırken bu veriler kullanılmaktadır. Gelişmiş ülkelerdeki bildirim oranlarına ulaşılması için hem sağlık mesleği mensupları hem de tüketiciler arasında bu konuda farkındalık yaratılmasına ihtiyaç vardır.

Ulusal farmakovijilans sisteminin paydaşları
Verimli bir farmakovijilans sisteminin işlerliği için tüm paydaşlara önemli sorumluluklar düşmektedir (3).
Ruhsat sahibi: Ruhsatına sahip olduğu ilaçların güvenliliğini garanti eder. Bu kapsamda, ilaçlarının güvenliliğini sürekli izlemekten, ilacın ruhsatlı olduğu diğer ülkelerin yetkili otoriteleri tarafından getirilen yasaklama ve kısıtlamalar da dahil olmak üzere ilacın yarar / risk değerlendirmesini etkileyebilecek herhangi bir değişiklik konusunda ürün bilgilerinin bilimsel veriler ışığında güncel tutulmasından sorumludur. Bu sorumluluk, ilacın kullanımının ruhsata dâhil olup olmadığından bağımsız olarak tüm popülasyon ve endikasyonlar için elde edilen pozitif ve negatif bulguları da kapsar. Farmakovijilans sistemine sahiptir; bu sistem vasıtasıyla riski en aza indirmek ve önlemek için gereken tedbirleri alır. İlaçlarıyla ilgili olarak meydana gelen advers reaksiyonların sınıflandırılmasında MedDRA terminolojisi kullanır. Ülkemize ait verileri de içeren tüm bilimsel ve tıbbi literatürü, şüpheli advers reaksiyon olgu raporları açısından önemli bir bilgi kaynağı olması nedeniyle izler ve Türkiye’de meydana gelen tüm ciddi advers reaksiyonları on beş gün içerisinde TÜFAM’a bildirir. Risk yönetim sistemini, ilaca ait tanımlanmış ve potansiyel risklerle, ayrıca ruhsatlandırma sonrası güvenlilik verilerinin gerektirdikleriyle orantılı olarak hazırlar ve gerektiğinde günceller; yeni riskler olup olmadığını, risklerin değişip değişmediğini veya ilacın yarar / risk dengesinde herhangi bir değişiklik olup olmadığını tespit etmek üzere farmakovijilans verilerini izler. Ek izlemeye tabi ilaç listesinde bulunan ilaçlar için kısa ürün bilgisine bir ters eşkenar siyah üçgen sembolü ve bu sembolü takiben standart bir metin ekler. Ayrıca, tüketicilerin şüpheli advers reaksiyonları sağlık mesleği mensuplarına veya doğrudan TÜFAM’a bildirmelerini isteyen standart metni ilacın kullanma talimatına ekler. Ruhsat sahibi kuruma sunulan farmakovijilans ile ilgili bilgi veya belgelerin doğruluğunu ve güncelliğini taahhüt eder ve sonuçlarından doğacak yasal sorumluluğu üstlenir (3).
Sağlık mesleği mensupları: Hastalarda ilaç kullanımına bağlı olarak ortaya çıkan advers reaksiyonların TÜFAM’a spontan bildirimi, advers reaksiyonları gözlemleyen sağlık mesleği mensubunun mesleki sorumluluğundadır. Bu bildirimler, yönetmelikte öngörülen şekilde gerçekleştirilir (3).
Sağlık kurum ve kuruluşları: Kuruluş içi farmakovijilans sistemini kurar ve yönetmeliğe göre faaliyet gösterir. TÜFAM’a bilgi akışını sağlamak üzere hastane yönetimi tarafından farmakovijilans irtibat noktası görevlendirir (3).
Kurum: Farmakovijilans sistemi vasıtasıyla tüm verileri bilimsel olarak değerlendirir, riski en aza indirme ve önleme seçeneklerini dikkate alır. Türkiye’de ortaya çıkan ve sağlık mesleği mensupları ve tüketiciler tarafından bildirilen şüpheli advers reaksiyonları kayıt altına alır ve Dünya Sağlık Örgütü İlaç İzleme İş Birliği Merkezi’ne gönderir. Şüpheli ciddi advers reaksiyonlara ilişkin raporu, bildirimini izleyen on beş gün içinde ruhsat sahibine iletmekle yükümlüdür. Ek izlemeye tabi ilaçların listesini uluslararası uygulamaları da göz önüne alarak hazırlar ve günceller. Risk yönetim planını değerlendirir, riski en aza indirmeye yönelik tedbirlerin sonuçlarını izler (3).
Kurum, ruhsatlı ilaçlarla ilgili olarak aşağıdaki hususları dikkate alır (3):
- Risk yönetim planı kapsamında, riski en aza indirmeye yönelik tedbirlerin sonuçlarını izlemek.
- Risk yönetim sisteminde yapılan güncellemeleri değerlendirmek.
- Yeni risklerin olup olmadığını veya bu risklerin değişip değişmediğini ya da bu risklerin yarar / risk dengesi üzerinde etkisi olup olmadığını tespit etmek üzere PYRDR de dahil gerekli değerlendirmeleri yapmak.
- Kurum ve ruhsat sahibi, yeni risklerin meydana gelmesi halinde veya yarar / risk dengesinde değişiklik olması durumunda birbirlerini bilgilendirir.
Yapılan değerlendirmenin ardından Kurum farmakovijilansla ilgili durumlarda aşağıdaki işlemlerden uygun olanları başlatır ve bunu ruhsat sahibine bildirir (3):
- Ruhsatlandırma sonrası güvenlilik çalışması yapılması.
- Riski en aza indirecek tedbirlerin uygulanması.
- Ruhsatın askıya alınması, iptali veya ruhsatın geçerlilik süresinin uzatılmasının reddi.
- İlaç tedariğinin yasaklanması.
- Yeni kontrendikasyon ilavesi, önerilen dozun azaltılması veya endikasyon kısıtlaması gibi ilaç bilgilerinde değişiklik.
Biyolojik ve Biyobenzer İlaçların Farmakovijilans Süreçleri
Biyolojik ve biyobenzer ilaçların farmakovijilans süreçlerinin tanımlanması büyük önem teşkil etmektedir. Zira, biyolojik etkin maddeler; güvenlilik, kalite ve etkililik profilini biçimlendiren birçok basamak içeren, karmaşık üretim işlemleri kullanılarak üretilen kompleks moleküllerdir ve tüm farmasötiklere göre iki kat daha hızlı büyüdükleri görülmektedir.
Biyofarmasötik pazarında mevcut birçok biyolojik ilacın patent süresinin dolması biyobenzer ilaçların üretimi konusunu ön plana çıkarmıştır. Biyobenzer ilaçlar patentleri sona eren mevcut biyofarmasötik ürünlerin yeni versiyonlarıdır ve çoğunlukla proteinlerden oluşur. Biyobenzer veya biyobenzerlik kavramı tam anlamıyla bir biyolojik ürünün referans ürününe klinik olarak inaktif komponentlerdeki minör farklılıklarına rağmen yüksek anlamda benzemesi demektir. Biyobenzer ilaçların ruhsatlandırılması için biyobenzer ilaç referans ilaca benzer olmalı ve referans ürün ile arasında nitelik, güvenlilik ve etkililik açısından önemli bir fark olmamalıdır (4).
Üretim işlemi, ürünün etkin maddesi ile aynı ölçüde kalitesinin belirleyicisidir ve herhangi bir üretim basamağında yapılacak küçük değişiklikler ürün kalitesini ve bunun sonucu olarak ürünün güvenliliğini ve etkililiğini değiştirebilmektedir.
Biyobenzerleri jenerik ürünlerden ayıran ve bu konu ile ilgili 2001 yılında ilk mevzuatı oluşturan sağlık otoritesi EMA’dır. Tüm dünyada yeni biyobenzer ürün geliştirme projeleri preklinik ve faz çalışmalarıyla devam etmekte ya da ruhsat onayını beklemektedir.
İmmünojenisite
Büyük moleküllü ve biyolojik bir ajanın vücuda enjekte edilmesi bağışıklık sistemini uyarır. Uyarılmış bağışıklık sistemi cilt döküntülerinden başlayıp, ölümcül kemik iliği süpresyonu ya da anafilaksi gibi ciddi klinik sonuçlar oluşturabilir. Ayrıca oluşan antikorlar da ilacın etkinliğini sona erdirir, dolayısıyla hastalar o ilaç sınıfına karşı direnç geliştirebilir.
İmmünojenisite potansiyel olarak klinik açıdan önemli olan ve ürüne özgü farmakovijilans ve risk yönetimi faaliyetleri gerektirebilecek istenmeyen bir bağışıklık cevabı ifade eder.
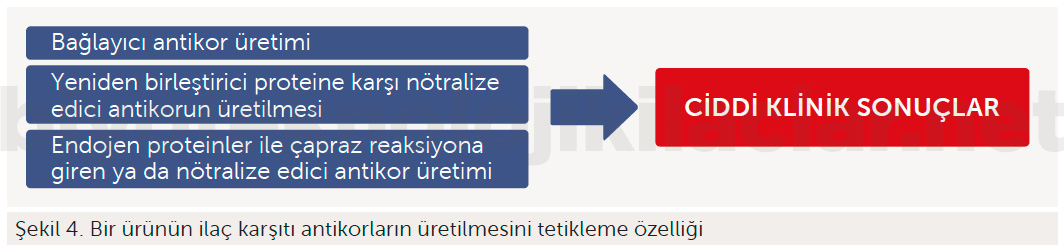
Örneğin, antieritropoietin antikorlarının doğal proteinleri nötralize etmesi ve progresif ve dirençli bir anemiye yol açması sebebiyle immünojenisite önemli bir sorundur.
Biyolojik ve biyobenzer ilaçların yapılarının büyük olması, yapılarının karmaşık olması, üretimlerinin son derece karmaşık olması, safsızlaştırma sorunları, stabilite sorunları (aktivite azalması, kolay bozulma), immünojenisite sorunları (alerjik reaksiyonlar, etkisizlik), formülasyon zorluğu, uygulama yollarının sınırlı olması (sadece parenteral) nedeniyle kullanımı sırasında hastada ortaya çıkabilecek her türlü olumsuz durum ve diğer yan etkiler (kilo problemleri, hipoglisemi gibi) fark edilmeli, kaydedilmeli ve bildirilmelidir.
Hastalarda biyolojik ve biyobenzer ilaçların kullanımına bağlı olarak ortaya çıkan ürün advers etkilerinin kuruma spontan bildirimi, advers etkileri gözlemleyen sağlık mesleği mensubunun mesleki sorumluluğunda olup; bu bildirimler, farmakovijilans yönetmeliğinde öngörülen şekilde gerçekleştirilmelidir.
Biyolojik bir ürünün güvenlilik profilini immünojenik özellikler dahil olmak üzere; etkin maddenin, yardımcı maddelerin, işlemle ilişkili impüritelerin direkt veya indirekt farmakolojik özellikleri, konakçıyla veya hastalıkla ilişkili duyarlılığı (örn; ilaç kaynaklı alerjik reaksiyonlar, oto-immünite ve inflamatuvar olaylar) belirlemektedir.
Tüm rekombinant protein ilaçlar hastanın immün sistemi tarafından yabancı olarak tanınma potansiyeline sahiptir. Ürüne çok defa maruz kalmanın antikor üretimini tetikleyebilmesi, antikorları tespit etmek için kullanılacak yöntemle ilgili kesin bir kanı olmaması, deneylerin geniş çapta farklılık göstermesi, antikorların ortaya çıkma süresinin uzun oluşu, hastaların eş zamanlı farklı ilaçlara maruz kalması, farklı dozlarda farklı endikasyonlarda ilaç uygulaması, farmakodinamik sürecin uzun olması, klinik olarak immünojenisiteye sebep olan etkenin tespitini ve dolayısıyla farmakovijilans aktivitelerini de zorlaştırmaktadır.
Bir biyolojik üründen diğerine geçmek antikor üretim riskini ciddi şekilde arttırabilir. Biyobenzer ilaç hasta immün sisteminden ve hastalıktan kaynaklanan özelliklerden referans üründen farklı etkilenebileceği de unutulmamalıdır. Bu nedenle, hekimlerin her zaman biyolojiklerin değiştirilmesi kararına müdahil edilmesi, eczane ikamesi (substitüsyon) için tedaviyi yapan hekimin önceden bilgili onayının alınması doğru farmakovijilans yönetimi için çok önemlidir (4-7).
Biyolojik ürünlere ilişkin immünojenisite kaynakları çok faktörlüdür:

İmmünojenisitenin olası klinik sonuçları (8)
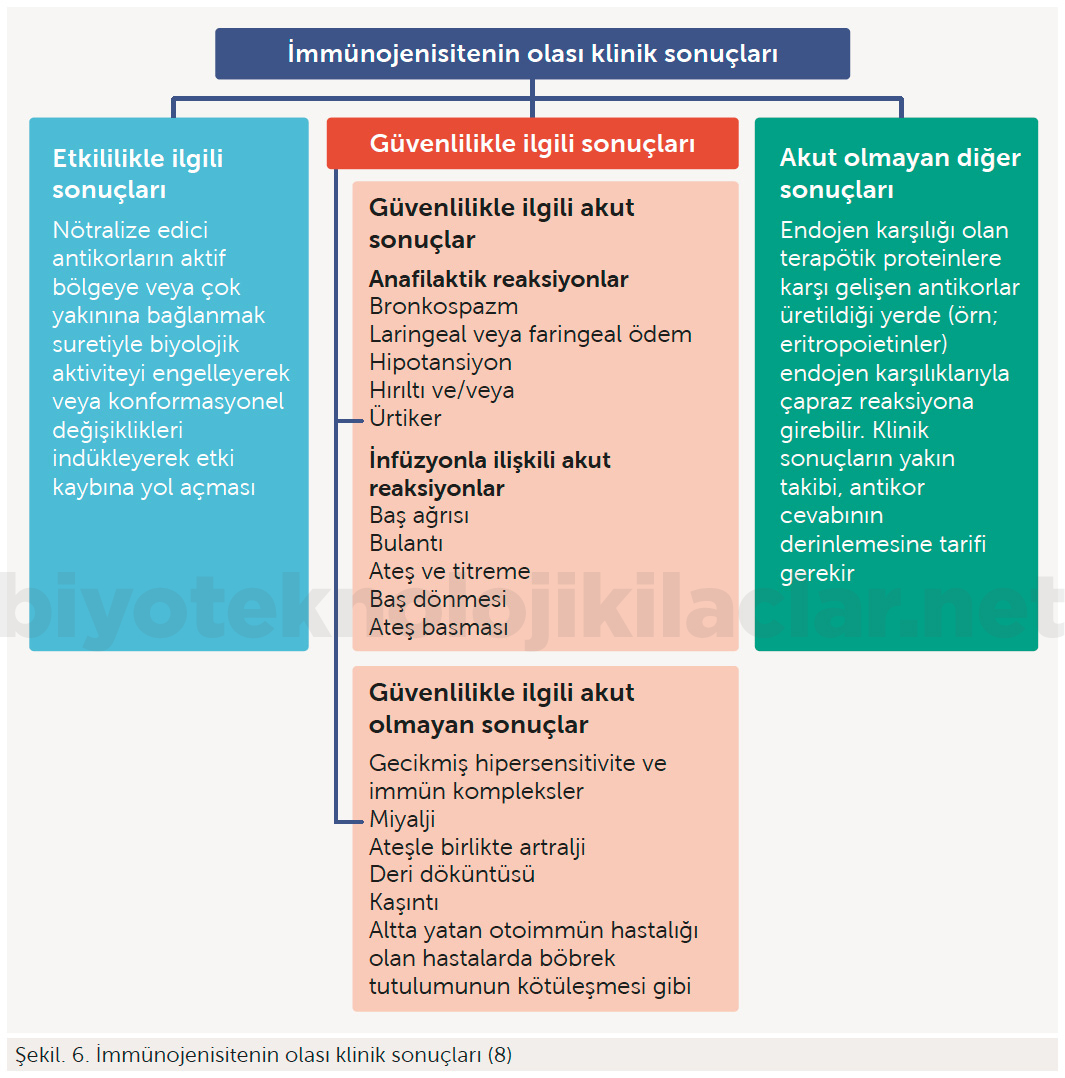
Etkililiğin azalması ve güvenlilik profilinin değişmesinin ilişkili olması zorunlu değildir. İnfüzyonla ilişkili reaksiyonlar gibi güvenlilik sorunları, etkililik kaybı olmadığında dahi ortaya çıkabilir.
Ruhsatlandırma öncesi immünojenisite değerlendirmesi
Biyoteknolojik ilaçların ürün geliştirme süreci esnasında ve ruhsatlandırmadan önce spesifik immünojenisite değerlendirmesi yapılması gerekmektedir. Ancak klinik olmayan modeller ve analitik / biyotayin yöntemleriyle insandaki immünojenisite öngörülemeyebilir.
Ruhsatlandırma öncesi çalışmalara ilişkin sınırlı örneklem büyüklüğü, tedavi edilecek hastalığın nadir görülmesi, nadir olarak meydana gelen immünojenisite durumlarının ruhsatlandırma öncesinde değerlendirilmesine olanak vermeyebilir (9).
Ruhsatlandırma sonrası immünojenisite değerlendirmesi
Ruhsatlandırma sonrası biyobenzer ürünlerin klinik güvenliliğinin devamlı ve yakın bir şekilde takip edilmesi ve sürekli bir yarar / risk değerlendirmesi yürütülmesi gerekmektedir (9).
İmmünojenisite konusunda yapılan çalışmalar ve pazarlama sonrası nasıl takip edileceği risk yönetim planında anlatılmalıdır (9).
Belirsizlik olması durumunda ruhsatlandırma sonrası spesifik faaliyetler ve gözetim çalışmalarının gerçekleştirilmesi gerektiği göz önünde bulundurulmalıdır (10).
Üretim Değişikliği
Ruhsat sahipleri, ruhsatlandırma sonrasında ürünlerinin üretim işlemlerinde sıklıkla değişiklik yapmaktadır. Üretim değişiklikleri biyolojik ürünlerde daha karmaşık olabilir.
Değişiklik öncesi ve sonrası ürünlerin kalite, güvenlilik ve etkililiğinin advers şekilde etkilenmeyecek ölçüde karşılaştırılabilir olduğunun belirlenmesi için bunların karşılaştırılabilirlik testleriyle desteklenmesi gerekmektedir (11).
Biyoteknoloji kaynaklı veya biyobenzer ruhsatlı ilaçlar için yeni üretim işlemi kullanılması durumunda risk yönetim planı sunulmalıdır.
Stabilite ve Soğuk Zincir
Üretim süreçlerinin ve standartlarının izin verilen spesifikasyon dahilinde kaldığından emin olunması için biyolojik ürünler konusunda sıkı süreç kontrolleri yürürlükte olmalıdır (12).
Bu süreçlere ve standartlara uyulmaması biyolojik ürünlerin stabilitesinde ve kalitesinde, biyolojik olmayan ürünlere kıyasla daha fazla etkiye neden olabilir ve bu durum immünojenisite ortaya çıkmasına veya immünojenisitede değişikliğe ya da kontaminasyona yol açabilir (13).
Adlandırma ve Ürünün İzlenebilirliği
Uluslararası ortak ad (INN): Etkin madde veya maddelerin uluslararası ve mülkiyete konu olmayan adıdır. INN sistemi hastalar için ilaçların net bir şekilde belirlenmesi, güvenli reçetelenmesi ve dağıtımı için ve dünya çapındaki sağlık sektörü çalışanları ve bilim insanlarının iletişimi ve bilgi alışverişi açısından önemlidir (14).
Ancak biyoteknoloji ürünleri için niteleyici ve kolay anlaşılır bir tanımlama yöntemi olmalıdır. Biyoteknolojik ürünlerde etkili bir farmakovijilans faaliyeti yürütmek için ürünün tanımlanması, güvenlilik profillerinin doğru biçimde kategorize edilmesi gerekir. Biyoteknolojik ürünlerin takip edilebilir olması için özgün kimlikleri olmalı ve uluslararası ortak adına (INN) ek olarak marka adı, üretici firmanın ismi, lot numarası ve üretildiği ülke bilgileri olmalıdır (14).
Uygulanan bir biyolojik ürünün adı ve seri numarası hastaya verilmeden önce sağlık mesleği mensubu tarafından dosyasına kaydedilmelidir. Ruhsatlandırma sonrası klinik kullanımda sürekli olarak ürün ve seri numarası izlenebilirliğinin sağlanması gerekmekte olup bu konuya risk yönetim planlarında yer verilmelidir.
Ek izleme
Ek izlem, bir pazarlama sonrası takip prosedürüdür. Bu prosedür, güvenlilik profili henüz tam olarak tanımlanmamış yeni ruhsatlandırılmış ilaçların veya tanımlanmasına ihtiyaç duyulan yeni güvenlilik sorunları olan ilaçların advers reaksiyon bildirim oranlarını artırma ihtiyacına dayanmaktadır (15).
Başlıca hedef, ilaçların klinik uygulamadaki risk profillerini daha da netleştirmek amacıyla ek bilgilerin mümkün olduğunca erken toplanmasını sağlamak ve böylelikle ilaçların güvenli ve etkili bir biçimde kullanılmasına bilgi desteği sağlamaktır (16).
Biyoteknolojik ilaçlar ek izleme tâbidir (15). İlaçların Güvenliliği Hakkında Yönetmelikte öngörülen şekilde bu ilaçların ters eşkenar siyah üçgen (▼) işaretiyle kolayca tanımlanması sağlanmaktadır. Üçgenin ardından kısa ürün bilgilerinde (KÜB) aşağıdaki açıklayıcı beyan yer almaktadır: “Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM’a bildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır?”. Kullanma talimatında da benzer bir ifade yer almalıdır. Bu açıklayıcı ifade sağlık mesleği mensuplarını ve hastaları tüm şüpheli advers reaksiyonları bildirmeye teşvik etme amacı taşımaktadır (17).
Şeffaf ve Anlaşılır KÜB ve KT Bilgisi
Hekimler, eczacılar ve hastaların ilacı destekleyen veri hususunda bilgi sahibi olmaları gerekir.
Kısa ürün bilgileri şeffaf olmalı ve aşağıdaki içermelidir:
- Biyolojik ve Biyobenzer İlaçlar ek izlemeye tabi ilaç listesindedir ve kısa ürün bilgilerinde ters eşkenar siyah üçgen sembolü yer almalıdır.
- Orijinal biyolojik veya biyobenzer ilaç olduğuna dair bilgi yer almalıdır.
- Ürünü biyobenzer olarak tanımlamalıdır.
- Referans ürünü tanımlamalıdır.
- Her bir endikasyonun nasıl onaylandığını tanımlamalıdır.
- Ürünün güvenlilik ve etkililik klinik verilerini tanımlamalıdır.
- KÜB, referans ürünle biyobenzer ürün arasındaki farkların bir özetini içermelidir
Risk Yönetim Planı
Risk Yönetim Planı (RYP), ilacın yararının risklerinden mümkün olduğunca daha fazla olmasını sağlamak amacıyla planlanan bir dizi farmakovijilans faaliyeti ve müdahalesidir. İlaçla ilgili riskleri tespit etmek, tanımlamak, önlemek, en aza indirmek için tasarlanır. İlacın ömrü boyunca devam eden bir süreçtir (18).
İlk ruhsat başvurusu sırasında, belirtilen endikasyon(lar)da risk / yarar dengesinin hedef popülasyon açısından olumlu olup olmadığı değerlendirilmektedir. İlacın gerçek ve potansiyel risklerinin tümü henüz tespit edilmiş olmayabilir. Ayrıca, birden fazla farklı riskin olabileceği hedef popülasyonun geneline kıyasla daha büyük olup etkilenen hasta alt kümelerinin de bulunabileceği ihtimali göz önüne alınır. Risk yönetimi kapsamında gerçekleştirilen faaliyetler teknik, bilimsel ve yasal gelişmelerin yanı sıra elde edilen yeni bilgilere, algılanan risklere, bunların halk sağlığı üzerindeki öngörülen etkilerine ve ürünün yaşam döngüsünün hangi evresinde bulunduğuna bağlı olarak değişiklik gösterebilir (18).
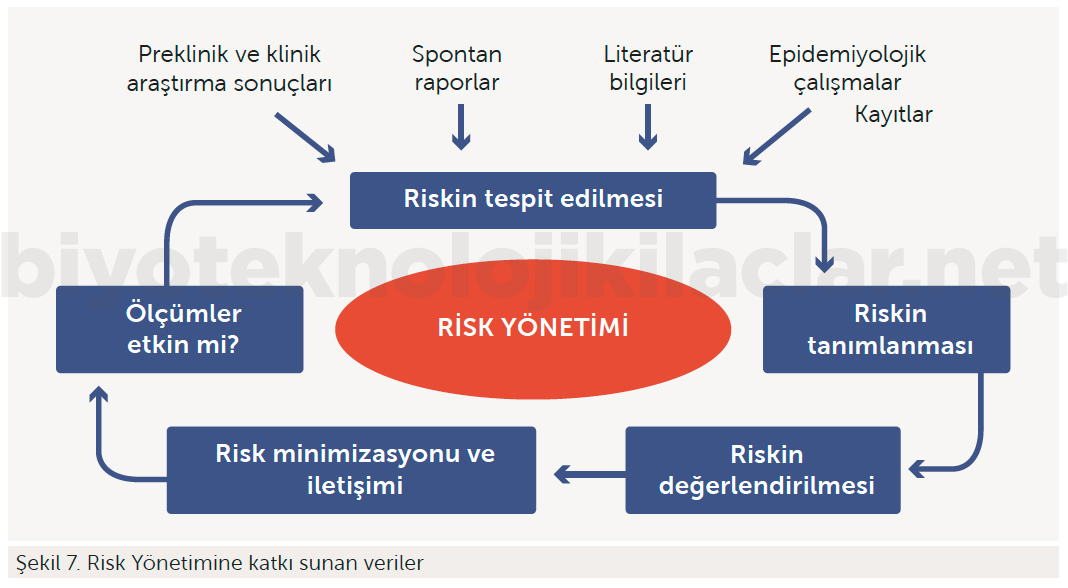
Risk yönetime katkı sunan veriler ve döngüsü Şekil 7’de gösterilmektedir.
Başvuru / ruhsat sahibi tarafından aşağıdaki durumlarda RYP sunulması gerekmektedir:

Bir riskin yönetilebilmesi için izlenen süreç dört basamaktan oluşmaktadır. Bu süreç; riskin tespiti, değerlendirilmesi, en aza indirilmesi ve iletişimini kapsar.
Bir Risk Yönetimi Planı hazırlanırken dikkate alınması gereken temel noktalar vardır.
- Güvenlilik spesifikasyonu oluşturulurken gerekli tüm veriler incelenmelidir. Ürün dosyasının diğer bölümlerinde yer alan ve güvenlilik spesifikasyonunda tartışılmayan (tamamlanmamış) hususlar olmamalıdır.
- Hedef popülasyona yönelik çalışılmayan bölümlerin neler olduğu ve potansiyel riskler / eksik bilgilere ilişkin uygun güvenlilik endişelerinin dahil edilmesi
- Güvenlilik veri tabanının kısıtlılıkları ve ilacın güvenlilik profiliyle ilgili olarak nasıl bir güvence sağlandığı bilgisi
- ICH-E2E kapsamında belirtilenler haricinde başka spesifik riskler mevcut mudur?(örn; endikasyon dışı kullanım, ilaç suistimali ve kötüye kullanımı, enfeksiyöz hastalıkların bulaşması, ilaç kullanım hatası, vb.)
- Güvenlilik spesifikasyonu ilaca ilişkin güvenlilik sorunlarının (örn; önemli tanımlanmış riskler, önemli potansiyel riskler ve eksik bilgiler) gerçek yansımasını içermelidir.
- Bilinen etkin maddenin daha önce başvurusu yapılmışsa, güvenlilik spesifikasyonuna referans ilaca ilişkin tüm güvenlilik endişeleri eklenmelidir.
- Tanımlandığı şekliyle terapötik araçlar arasındaki yeri, hedeflenen endikasyon ve mevcut tıbbi uygulamayla uyumu
- Güvenlilik spesifikasyonundaki tüm güvenlilik endişeleri farmakovijilans planı kapsamına alınmalıdır.
- Rutin farmakovijilans faaliyetleri yeterli midir yoksa ilave farmakovijilans faaliyetlerinin gerekli olup olmadığı tartışılmalıdır.
- Risk minimizasyon tedbirlerinin neler olduğu ve risk minimizasyon faaliyetlerinin etkinliğinin değerlendirilmesi yapılmalıdır.
- Eğer ilaç kullanım hatası bir güvenlilik endişesi oluşturuyorsa, RYP bunların izlenmesine ilişkin gerekli önerileri içermelidir.
- Ek çalışmalar gerekli midir? Farmakovijilans planında önerilen çalışmalar bilimsel soruların cevaplanması için yeterli midir ve bu çalışmalar gerçekleştirilebilir nitelikte midir?
- Önerilen faaliyetler, bu faaliyetlerin sonuçlarının sunulması ve farmakovijilans planının güncellenmesine ilişkin gerekli süreler ve dönüm noktaları tanımlanmalıdır.
- Etkililiğe ilişkin ruhsatlandırma sonrası çalışmalar gerektiğinde plan sunulmalıdır.
- İlacın etkililiğine ilişkin tanım ve bunların dayandırıldığı çalışmalar ve sonlanma noktaları dosyanın içeriği ile uyumlu olmalıdır.
- Ürün bilgisi tanımlanmış tüm önemli riskleri ve eksik bilgileri uygun şekilde yansıtmalıdır.
- Ürün bilgilerinde riskler için önerilen ifadeler ve bunların konumu uygun ve ilgili kılavuzlarla uyumlu olmalıdır (örn; KÜB kılavuzu).
- Ruhsat sahibi ilaç kullanım hatalarını azaltmaya yönelik yolları göz önünde bulundurmalıdır (geçerli olduğunda cihaz tasarımı, ambalaj tasarımı gibi).
- Risk minimizasyon faaliyetlerinin etkinliğinin ölçülmesi ve değerlendirilmesine yönelik yöntemler uygun ve iyi tanımlanmış olmalıdır.
Biyolojik ve Biyobenzer İlaçların farmakovijilans planı güvenlilik sorunlarını tespit ve tanımlamaya yönelik tedbirler, riskleri azaltmaya ya da önlemeye yönelik faaliyetlerin risk yönetim planınında yer almalıdır. Biyobenzer ürünlerin risk yönetim planlarını hazırlayabilmek için bu verilerin ürüne özgü olarak oluşturulması gerekmektedir. Risk Yönetim Planının hazırlanması ve sunumuna yönelik format, ilgili “İyi Farmakovijilans Uygulamaları” kılavuzu Modül 6 – Risk Yönetim Sistemleri”nde yer almaktadır.
Risk Yönetim Planı aşağıda gösterildiği gibi iki temel bölümden oluşur (19)

I. Bölüm
Güvenlilik Spesifikasyonu: Güvenlilik Spesifikasyonu bölümünde ilaca ilişkin tespit edilen önemli riskler, önemli potansiyel riskler ve önemli bilgi eksiklikleri özetlenmelidir. Potansiyel olarak risk altındaki popülasyonları (ürünün kullanılması muhtemel olanlar) ve henüz çözümlenememiş olan ve risk / yarar profilini daha iyi anlamak için ruhsat sonrası dönemde daha fazla araştırma yürütmeyi gerektiren güvenlilik sorunları da ele alınmalıdır. Güvenlilik Spesifikasyonu özel bir veri toplama ihtiyacı olup olmadığını tespit etme konusunda yardımcı olma ve Farmakovijilans Planını oluşturmayı kolaylaştırma amacını taşır.
Farmakovijilans Planı: Bu bölüm daima güvenlilik spesifikasyonunu temel almalı ve tespit edilen güvenlilik sorunlarına yönelik tedbir önerilerinde bulunmalıdır. Özel bir sorun yaşanmayan ilaçlarda ruhsat sonrası güvenlilik izleme çalışmaları kapsamında ilave bir tedbire (örneğin, güvenlilik çalışmaları) gerek olmaksızın yalnızca rutin farmakovijilans faaliyeti yürütmek yeterli olacaktır.
II. Bölüm
Risk minimizasyonu faaliyetlerine duyulan ihtiyaç ile ilgili bir değerlendirme: Tespit edilmiş önemli risklerin, önemli potansiyel risklerin ya da önemli bilgi eksikliğinin olduğu ilaçlarda güvenlilik sorunlarını ele almaya yönelik ilave farmakovijilans faaliyetlerinin tasarlanması düşünülmelidir (epidemiyolojik çalışmalar, yoğun izlem gibi). Önerilen risk minimizasyon faaliyetlerinin konusu olan güvenlilik sorunları risk minimizasyon planında liste halinde verilmelidir. Planda hem rutin hem de ilave risk minimizasyon planlarına yer verilmelidir.
Risk minimizasyon faaliyetleri: Önerilen ilave risk minimizasyon faaliyetlerinin neler olduğu, her biri için tedbirin riski azaltmadaki etkinliğinin nasıl değerlendirileceği detaylı olarak anlatılmalıdır.
Risklerin minimize edilmesi için alınabilecek önlemler;
✓ Bilgi temini: Bir ürünle ilgili riskler ve risklerin minimize edilmesi için alınması gereken önlemler arasında birincisi ve belki de en önemlisi sağlık mesleği mensuplarının ve hastaların bilgilendirilmesidir. Bilgilendirme, KÜB ve KT içinde yer alan bilgilerle sınırlı olabilir, ilave eğitim materyalleri kullanılabilir ya da ürünlerin dış ambalajı üzerine uyarı metinleri yazılabilir. KÜB’lere çerçeve içinde uyarı konularak sağlık mesleği mensuplarının ilacın riskleri konusunda uyarılması sağlanabilir.
✓ Eğitim materyali: İlacın güvenliliği ile ilgili olarak meydana gelen gelişmelerden sağlık mesleği mensuplarını ivedilikle haberdar edebilmek için bilgilendirme mektupları dağıtılmalıdır. Piyasaya yeni çıkan ve potansiyel riskleri nedeniyle takip edilmesi gereken ilaçlar için TÜFAM’ın onayı ile hekimlere ve hastalara yönelik broşürler dağıtılmaktadır. Bu uygulama pek çok ilaca uygulanmıştır (Ek-1 ve Ek-2). Besinlerle etkileşmesi ile ilgili riski olan ilaçlar için varfarin örneğinde olduğu gibi hastaların bilgilendirilmesine yönelik tedavi rehberi hazırlanmalıdır. Ülkemizde henüz ruhsatlı olmayan, ancak endikasyon dışı olarak Bakanlığın izniyle kullanılabilen ilaçlar için örneğin aşılarda, hekim aşı sertifikası doldurulmalı ve hastalara hasta bilgilendirme kartı dağıtılmalıdır.
✓ İlacın yasal statüsü: Bir ilacı reçete etmesine izin verilenlerin kimler olacağı ya da bir hastanın ilacı hangi koşullarda alabileceği kontrol altında olmalıdır. İlacın yasal statüsüyle ilgili bir başka konu da kısıtlı reçete uygulamasıdır. 2011 yılında kısıtlı reçete uygulamasına geçilmiştir.
✓ Eczane seviyesinde kontrol: Eczacıların ilaçların riskleri konusunda eğitilmesi ve bilinçlenmesi hastaların eğitilmesine katkı sağlayacaktır.
✓ Reçete sayısının ve geçerliliğinin kontrolü: Reçete edilen kutu sayısının sınırlandırılması da risk minimizasyon faaliyetidir. Reçete edilen kutu sayısı sınırlanarak, hastanın sağlık mesleği mensubunu belli aralıklarla görmesi risklerin minimize edilmesi için alınması gereken önlemlerdir.
✓ Bilgilendirilmiş gönüllü oluru ve hastayla ilgili diğer hususlar: Hastalara ilacın risklerini anlatmak ve bu riskleri azaltmak için uygulanacak önlemlerle ilgili bilgi verildiğini garanti altına almak açısından hasta onay formu kullanımı bir risk yönetimi etkinliğidir. Ülkemizde bazı biyolojik ilaçların tanımlanmış ve potansiyel riskleri nedeniyle, bu ilaçlar için hazırlanan hasta onay formları kullanılmaktadır (Ek-1).
✓ Sınırlı erişim programları
✓ Hasta kayıtları
Farmakovijilansın Beklentisi
• Ülkemizde ve dünyada, karşılanmamış tedavi ihtiyaçlarına yönelik yeni biyolojik ürünlerin keşfedilmesi ve kullanılmasında artış yaşanmaktadır. Bu artışla beraber dünya çapında biyobenzer ürünlerin sayısı da artmaktadır.
• Biyolojik ürün gruplarında kritik önem taşıyan immünojenisite, ekstrapolasyon, değiştirilebilirlik ve farmakovijilans planları gibi konuların biyobenzer ilaç grupları için de özenle ele alınması gerekmektedir.
• Biyobenzer ürünlerle ilgili etkililik ve güvenlilik takibi için sağlık profesyonellerinin ve hastaların bilgi sahibi olması gerekmektedir. Bu amaçla, biyobenzer ürünler için immünojenisite verilerini de içeren kontrollü klinik çalışmalar yapılmalıdır.
• Her biyobenzer ilacın kendine özgü bir risk yönetim planı oluşturulmalıdır. Bu ürünler için hasta izleme sistemlerinin takip edilebilir olması, advers olay bildirimlerinin artırılması, “risk penceresi” içindeki tüm immünojenik raporların ruhsat onayı öncesi ve sonrasında dikkatli bir şekilde tıbbi olarak değerlendirilmesi oldukça önemlidir.
Farmakovijilans uygulamaları sayesinde klinik kullanımı olan ruhsatlı / izinli ilaçların güvenlilik profillerini izleyerek, yeni güvenlik sinyallerine karşı erken önlemler alarak riski engelleme şansına sahibiz. Günümüzde ilaç üretiminde ve kullanımında yaşanan artış, ilaçlar hakkındaki bilgilerin sürekli güncellenmesi, yeni moleküller ve yeni endikasyonlar, ilaçlar hakkında bilinenlere yeni eklenenler ülkelerin ortak sorunları olup her ülkenin kendine has şartları, bu sorunların ortaya çıkış şeklini, şiddetini, sonuçlarını etkilemekte ve ulusal farmakovijilans sistemini gerekli kılmaktadır. Pazarlama sonrası, klinik kullanımda olan ilaçların risk / yarar oranlarının bireysel ve toplumsal düzeyde mümkün olabilecek en iyi duruma getirilmesini sağlamak farmakovijilansın görevidir.
Küçük moleküllü ilaçlarla karşılaştırıldığında, biyolojik ürünlerin karmaşıklığı, üretim süreçlerine bağlı ve yapısal değişkenliğe daha duyarlı olmaları, bağışıklıkla ilgili reaksiyonları indükleme potansiyeline sahip olmaları, bu ürünlerin güvenliliğinin izlenmesinin önemini daha da arttırmaktadır. Dolayısıyla, biyoteknolojik ilaçların pazara girmeden önce farmakovijilans ve risk yönetim planları oluşturulmalı, ürünün klinik uygulamada olduğu dönemde ise sürekli takibi yapılmalıdır.
Başvuru sahibi firmanın biyoteknolojik ürünler için oluşturduğu farmakovijilans sistemine önem vermesi, işlerlik kazandırması ve takip etmesi gereklidir. Ayrıca, ülke sağlık otoriteleri, sağlık çalışanları ve hastaların tüm biyoteknolojik ilaçların kullanımı sırasında karşılaşılan advers olay bildirimlerini takip etmesi büyük önem arz etmektedir. Farmakovijilans planı, güvenlilik spesifikasyonunu temel almalı ve tespit edilen güvenlilik sorunlarına yönelik tedbir önerilerinde bulunmalıdır.
Küçük moleküllü ürünlerde özel bir sorun yaşanmadığı takdirde ruhsatlandırma sonrası güvenlilik izleme çalışmaları kapsamında ilave bir tedbire gerek olmaksızın yalnızca rutin farmakovijilans faaliyeti yürütmek yeterli olduğu halde biyoteknolojik ürünler için ilave çalışmalar yapılması ve rutinde takip edilmesi için ürünlerin tanımlanabilir olması gerekir. Avrupa Birliği’nde biyolojik ürünlere yönelik farmakovijilansın iyileştirmelere ihtiyacı olduğu da raporlanmaktadır (20). Ürün preklinik, in vitro ve in vivo çalışmalarla değerlendirilmiş, toksisite açısından oluşabilecek farklıkları tespit etmek için yeterli sürede farklı doz uygulayarak toksikoloji çalışmaları yapılmış olmalıdır. Klinik etkililiğin karşılaştırılması amacıyla klinik farmakokinetik ve farmakodinamik çalışmalar ve takiben 2 veya 3 kollu klinik etkililik çalışmalarına sahip olması beklenir. Klinik güvenlilik açısından yan etki profili ve immünojenisite verileri gerekir. Ek immünojenisite verisi toplayabilmek için alınan önlemlere farmakovijilans planında yer verilmelidir. Referans ürünle gözlenen belli ciddi advers olaylara karşı ayrıntılı bilgi sunulmalıdır. Dolayısıyla başvuru dosyasında kapsamlı fizikokimyasal ve biyolojik karakterizasyon ile etkililik ve güvenlilik karşılaştırma çalışmaları yer almalıdır. Nitekim, Gıda ve İlaç İdaresi de (FDA) biyolojik ürünler için ana dosyalarla ilgili yönetmeliklerini değiştirmeyi önermektedir ve konuyla ilgili olarak 28 Haziran 2019’da web sayfasını 57 günlük bir süre (08/27/2019) tanımlayarak resmi yorumlara açmıştır (21).
Klinik uygulamada var olan biyolojik ilaçlarla ilgili şüpheli advers reaksiyonlar için doğası gereği ilgili ürünün kesin olarak tanımlanması özel önem taşımaktadır. Bu nedenle, ürünün adının ve seri numarasının açık olarak belirtilmesi gerekmektedir. Ortaya çıkan advers reaksiyonun tedavisinde kullanılan ilaçlar ve varsa diğer tedavi yöntemleri tarihleriyle birlikte yazılmalıdır. Advers reaksiyonun tedavisi için kullanılan ilaçlar eş zamanlı kullanım olarak değerlendirilmemelidir. Sınırlı klinik programda sık gözlenmeyen advers olaylar ve immünojenisite sorunları ürün pazara sunulmadan önce saptanamaz. Dolayısıyla, bu grup ilaçların pazara verilmesi sonrasında sürekli olarak güvenlilik ve immünojenisite verilerinin toplanması için bir risk yönetim planı sağlaması gerekmektedir.
TANIMLAR
Advers reaksiyon, Advers ilaç reaksiyonu (AİR), Şüpheli advers (ilaç) reaksiyon, Advers etki, İstenmeyen etki
Bir ilaca karşı gelişen zararlı ve amaçlanmayan cevaptır. Bu bağlamda ‘cevap’ terimi, ilaç ile advers olay arasında nedensellik ilişkisi bulunduğuna dair en azından makul bir olasılık bulunduğu anlamını taşımaktadır. Ürünün ruhsat şartları dahilinde ya da dışında kullanımından kaynaklanan ya da mesleki maruziyet sonucu ortaya çıkan advers reaksiyonlardır. İlacın ruhsat dışı kullanımı; endikasyon dışı kullanımı, aşırı doz kullanımı, kötüye kullanımı, suistimali ve kullanım hatalarını kapsamaktadır.
Asgari raporlanabilirlik kriterleri
Şüpheli advers reaksiyon olgularının bildirilmesi bağlamında, bir olguya ilişkin bildirilmesi gereken asgari veriler şunlardır:
- tanımlanabilir bir raportör,
- tanımlanabilir bir hasta,
- bir advers reaksiyon ve
- şüpheli ilaç.
Beklenmeyen advers reaksiyon
İlaca ait kısa ürün bilgileriyle nitelik, şiddet veya sonlanım açısından uyumlu olmayan advers reaksiyondur. Kısa ürün bilgilerinde (KÜB) belirtilen, ancak bu ürünle oluştuğu özellikle tanımlanmış olmayan sınıfla ilgili reaksiyonları da kapsamaktadır.
Ciddi advers reaksiyon
Ciddi advers reaksiyon ölüme, hayati tehlikeye, hastaneye yatmaya veya hastanede kalma süresinin uzamasına, kalıcı veya önemli bir sakatlığa veya maluliyete, doğumsal anomaliye veya kusura neden olan advers reaksiyonlardır. Bir olgunun ciddiyetini belirlemek için advers reaksiyon sırasındaki özellikler / sonuçlar dikkate alınmalıdır. Örneğin, “yaşamı tehdit edici” ifadesi, reaksiyon meydana geldiği sırada hastanın ölüm riski altında olduğu bir reaksiyon anlamına gelir; daha şiddetli olsaydı ölüme neden olabilecek olan bir reaksiyon bu tanıma dahil değildir. Diğer durumların ciddi reaksiyonlar olarak kabul edilip edilmeyeceğine karar verilirken tıbbi ve bilimsel yargıya başvurulmalıdır. Bazı tıbbi olaylar, hastayı tehlikeye atabilir ya da yukarıdaki sonuçlardan birinin önlenmesi için bir müdahale gerektirebilir. Bu tür önemli tıbbi olaylar “ciddi” olarak kabul edilmelidir. Bu durumlara örnek olarak; alerjik bronkospazm için acil serviste ya da evde yoğun tedavi görülmesi, hastaneye yatışa neden olmayan kan diskrazileri, konvülsiyonlar ya da ilaca bağımlılık gelişmesi veya ilacın suistimal edilmesi verilebilir. Bulaşıcı bir ajanın bir ilaç yoluyla bulaştığı şüphesi de ciddi advers reaksiyon olarak değerlendirilmektedir.
Ek izlemeye tabi ilaçlar, onaylı endikasyonlarda belirli bir hedef popülasyonda yarar / risk dengesinin ruhsat verildiği tarihte pozitif olduğu kabulüyle ruhsatlandırılır. Ancak tüm riskler ilk ruhsatlandırma sırasında tespit edilemez ve bir ilacın kullanımıyla ilişkilendirilen risklerin bazıları sadece ilacın yaşam döngüsünün ruhsat sonrası döneminde tespit edilebilir veya daha detaylı bir biçimde tanımlanması mümkün olabilir. Bu nedenle; ilaçların güvenlilik izlemini pekiştirmek amacıyla, belli ilaçlarda ek izleme kavramını da içine alan, ruhsat sonrası dönemde riskle orantılı veri toplanmasına yönelik bir yapı uygulamaya konulmuştur. Ek izleme kavramı, esas olarak güvenlilik profili henüz tam olarak tanımlanmamış yeni ruhsatlandırılmış ilaçların veya tanımlanmasına ihtiyaç duyulan yeni güvenlilik sorunları olan ilaçların advers reaksiyon bildirim oranlarını artırma ihtiyacına dayanmaktadır. Başlıca hedef, ilaçların klinik uygulamadaki risk profillerini daha da netleştirmek amacıyla ek bilgilerin mümkün olduğunca erken toplanmasını sağlamak ve böylelikle ilaçların güvenli ve etkili bir biçimde kullanılmasına bilgi desteği sağlamaktır.
Farmakovijilans
Advers reaksiyonların ve ilaçla ilgili diğer sorunların tespit edilmesi, değerlendirilmesi, anlaşılması ve önlenmesine yönelik yürütülen faaliyetler ve bilimsel çalışmalardır. Bu genel tanıma uygun olarak, ilgili düzenlemelere göre farmakovijilans faaliyetlerinin dayandırılması gereken ana amaçlar şunlardır: Ruhsatlı ilaçların ruhsat şartları dahilinde ya da haricinde kullanılmasından kaynaklanan ya da mesleki maruziyet nedeniyle ortaya çıkan advers reaksiyonların zararlarının önlenmesi. Özellikle, ilaçların güvenliliği hakkında hastalara, sağlık mesleği mensuplarına ve kamuoyuna zamanında bilgiler sunmak yoluyla, ilaçların güvenli ve etkili bir biçimde kullanılmasının teşvik edilmesi. Dolayısıyla, farmakovijilans hastaların ve halk sağlığının korunmasına katkıda bulunan bir faaliyettir.
Farmakovijilans irtibat noktası
Görev yaptığı sağlık kuruluşunda advers reaksiyonların bildirilmesini teşvik etmekten, eğitim ve bilgilendirme çalışmaları yapmaktan, kendisine ulaşan advers reaksiyon bildirimlerini TÜFAM’a iletmekten sorumlu hekim, eczacı, bunların bulunmadığı yerlerde diş hekimi.
İlacın kötüye kullanılması
Bir ilacın kasten ve uygun olmayan bir şekilde onaylı ürün bilgilerine aykırı kullanılmasıdır.
İlaç suistimali
Zararlı fiziksel veya psikolojik etkilerin eşlik ettiği, sürekli veya aralıklı olarak kasıtlı aşırı ilaç kullanımıdır.
Risk minimizasyon faaliyeti, risk minimizasyon tedbiri, riski en aza indirme faaliyeti
İlaca maruz kalınmasıyla ilişkilendirilen bir advers reaksiyonun ortaya çıkmasını önleme, meydana gelme olasılığını azaltma ya da oluşursa, şiddetini azaltma amacı taşıyan halk sağlığı müdahaleleridir. Bu faaliyetler rutin risk minimizasyon faaliyetlerinden (örn; ürün bilgisi gibi) ya da ilave risk minimizasyon faaliyetlerinden (örn; sağlık mesleği mensuplarına yönelik bilgilendirme mektupları / eğitici materyaller) oluşabilir.
Risk minimizasyonu: Bir advers etkinin oluşma olasılığını ya da oluşması halinde şiddetini azaltmaya yönelik bir dizi aktivitedir.
Risk yönetim sistemi: Risk yönetim sistemi, ilaçlarla ilgili riskleri tespit etmeye, tanımlamaya, önlemeye ve minimize etmeye yönelik bir dizi farmakovijilans faaliyet ve müdahalesi olup, bu müdahalelerin etkinliğinin değerlendirilmesini de içerir.
Rutin farmakovijilans: Beşeri İlaçların Güvenliğinin İzlenmesi ve Değerlendirilmesi Hakkında Yönetmelik gereğince tüm ilaçlar için uygulanması zorunlu olan farmakovijilans faaliyetleridir.
Rutin risk minimizasyon faaliyetleri: Bir advers etkinin oluşma olasılığı ya da oluşması halinde şiddetini azaltmak amacıyla Kısa Ürün Bilgilerinde ve Kullanma Talimatında yer verilen bilgi ve uyarılarla etiket ve ambalajın dikkatli kullanılmasıdır.
Tespit edilen risk: İlgi konusu ilaçla ilişkili olduğuna dair yeterli kanıt bulunan olumsuz olaylardır. Örneğin: Preklinik çalışmalarda yeterli ölçüde gösterilen ve klinik verilerle doğrulanan advers etkiler, iyi tasarlanmış klinik araştırmalarda ya da epidemiyolojik çalışmalarda gözlenen, karşılaştırma grubuyla (plasebo ya da etkin madde ya da ilaç verilmeyen grup) karşılaştırıldığında, ilgi konusu bir parametrede tespit edilen farkın nedensellik ilişkisini düşündürecek kadar büyük olduğu advers etkiler, anafilaktik reaksiyonlar ya da uygulama yeri reaksiyonları gibi, iyi belgelenmiş spontan bildirimlerde öne sürülen ve nedensellik ilişkisinin zamanlamanın örtüşmesiyle ve biyolojik olabilirlikle güçlü biçimde desteklendiği advers etkiler.
Sağlık mesleği mensubu
Şüpheli advers reaksiyonların bildirilmesi bağlamında; hekim, eczacı, diş hekimi, hemşire ve ebeler.
Sinyal
Gözlemler ve deneyler de dahil olmak üzere bir veya birden fazla kaynaktan alınan, bir müdahale ile bir veya birden fazla olay arasında olumlu ya da olumsuz olası yeni bir nedensellik ilişkisi bulunduğunu ya da bilinen bir nedensellik ilişkisinin yeni bir boyut kazandığını düşündüren ve doğrulayıcı işlem gerektiren bilgilerdir.
Spontan bildirim, Spontan rapor
Bir sağlık mesleği mensubu ya da tüketici tarafından Kuruma ya da ruhsat sahibine özellikle talep edilmeden iletilen, bir veya birden fazla ilaç verilen bir hastada oluşan bir veya birden fazla advers reaksiyonun tarif edildiği ve bir çalışmadan ya da organize veri toplama programından kaynaklanmayan bilgilerdir.
Türkiye Farmakovijilans Merkezi (TÜFAM)
Sağlık Bakanlığı, Türkiye İlaç ve Tıbbi Cihaz Kurumu, Farmakovijilans ve Kontrole Tabi Maddeler Dairesi bünyesinde yer almakta olan ve ülkemizde sağlık mesleği mensuplarından, hastalardan ve ruhsat sahiplerinden spontan advers reaksiyonların toplanmasından sorumlu tek yetkili devlet kuruluşudur.
Bildirilmesi gereken advers reaksiyonlar
- Ek izleme tabi ilaçlarla görülen tüm şüpheli advers reaksiyonlar a. Ek İzleme tabi ilaçların listesi https://www.titck.gov.tr ana sayfada “Önemli Belgeler” bölümünde “Ek İzlemeye Tabi İlaçlar” adı altında iki ayda bir ilan edilmektedir.
- Bir ilacın ne kadar süredir piyasada olduğuna bakılmaksızın ciddi advers reaksiyonlar.
- Beklenmeyen advers reaksiyonlar.
- Sıklığında artış meydana gelen advers reaksiyonlar.
- Biyolojik ilaçlar ve aşılar.
- Bitkisel ürünlerle meydana gelen advers reaksiyonlar.
- Gecikmiş ilaç etkileri.
- Gebelik ve emzirme gibi özel durumlar sırasında ilaç kullanımı.
- Pediyatrik ya da yaşlı popülasyonda ilaç kullanımı.
- Doz aşımı, suistimal, endikasyon dışı kullanım, kötüye kullanım, ilaç kullanım hatası ya da mesleki maruziyet ile ilgili şüpheli advers reaksiyonlar.
- Etkisizlik.
- Kalite kusurları veya sahte ilaçlarla ilgili şüpheli advers reaksiyonlar.
- İlaç yoluyla bir enfeksiyöz ajanın bulaşmasından şüphelenildiği durumlardır.